.png)
Tools
A lot of our work results in software tools and datasets that might be of interest to you. Feedback always welcome!
Featured
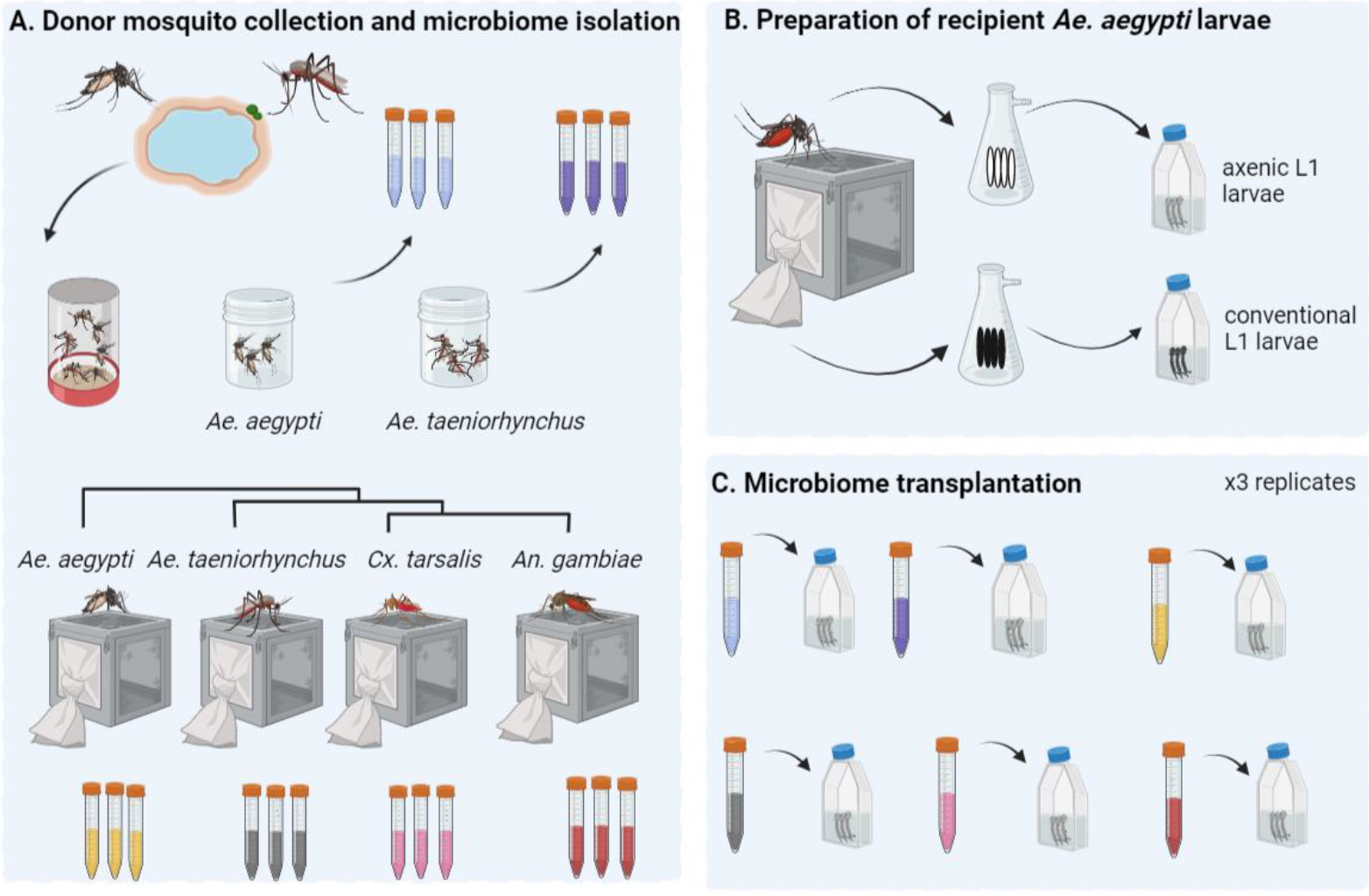 Microbiome-swap RNAseqAnalysis of transcriptome data to understand how a mosquito responds to a microbiome transplant
Microbiome-swap RNAseqAnalysis of transcriptome data to understand how a mosquito responds to a microbiome transplantUsing microbiome transplant experiments, we asked whether there were differences in transcriptional responses when mosquitoes of different species were used as microbiome donors. We used microbiomes from four different donor species spanning the phylogenetic breadth of the Culicidae, collected either from the laboratory or field. The repository provides the code used to run the analyses (R) and the raw counts table.
 MINUURMInUUR - Microbial INsight Using Unmapped Reads
MINUURMInUUR - Microbial INsight Using Unmapped ReadsMInUUR is a snakemake pipeline to extract unmapped whole genome shotgun sequencing reads and utilise a range of metagenomic analyses to characterise host-associated microbes. Orginally, MInUUR was intended to be used for the extraction of mosquito-associated bacterial symbionts, however, its application can be applied to other host-associated WGS data. MInUUR aims to leverage pre-existing WGS data to ‘scavenge’ for microbial information pertaining to host associated microbiomes - the key advantage being metagenomic reads as inputs to produce genus & species level classifications, functional inference and assembly of metagenome assembled genomes (MAGs).
More
 E. coli genome resourceA highly quality-controlled collection of E. coli genomes ready for species-wide analyses.
E. coli genome resourceA highly quality-controlled collection of E. coli genomes ready for species-wide analyses.This is a fantastic collection E. coli genomes by my previous PhD student, Gal Horesh, whom I was co-supervising whilst I was staff in the team of Nick Thomson at the Wellcome Sanger Institute. We hope that saves anyone interested a huge amount of time by being able to dive right into analyses rather than sorting out a genome collection!
 Pangenome classification
Pangenome classificationAn analysis workflow to increase the granularity of the accessory genome by taking different evolutionary trends into account; by my previous PhD student, Gal Horesh, whom I was co-supervising whilst I was staff in the team of Nick Thomson at the Wellcome Sanger Institute.
 mGEMS
mGEMSThis is a project led by Tommi Maeklin, who is a PhD student working with Antti Honkela at the University of Helsinki and Jukka Corander at the University of Oslo. This can be used to analyse a mix of (ideally pre-enriched) different strains of the same species and disentangle their genomes.
 rPinecone
rPineconeThis tool was developed by Alex Wailan whilst we were both working in the team of Nick Thomson at the Wellcome Sanger Institute. This R package defines the sub-lineages of a clonal expansion via a phylogenetic tree using a root-to-tip approach.